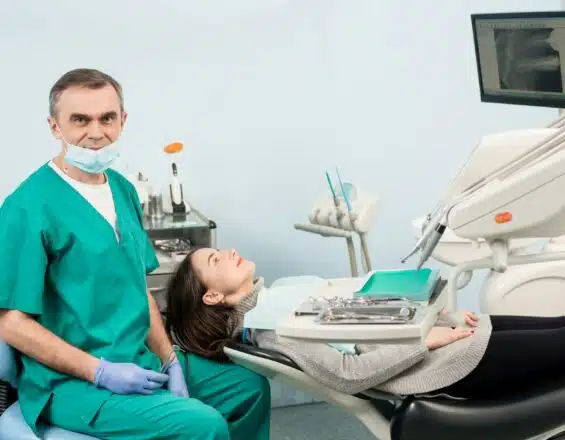



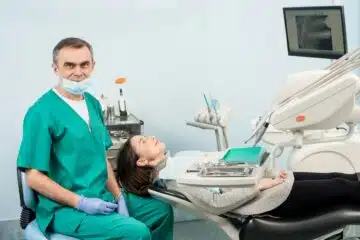
La santé dentaire est un aspect crucial du bien-être général, et lorsqu’une urgence dentaire survient, il est vivement recommandé de recevoir des soins appropriés rapidement pour atténuer la douleur et prévenir d’autres complications. Le chirurgien-dentiste est un professionnel de la santé spécialisé dans le traitement des affections dentaires, et il offre une gamme de solutions […]
Actu


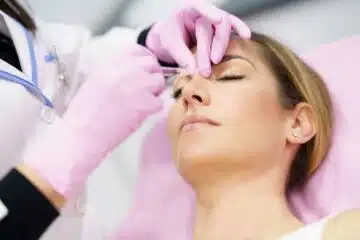
La rhinoplastie ultrasonique fait partie de la chirurgie plastique et esthétique. Des techniques évolutives qui s’affinent et constamment innovées pour des résultats meilleurs esthétiquement : en beauté, naturel, en harmonie, durables et respectant le côté fonctionnel de l’organe. Le Dr Rani Makhoul s’est spécialisé dans la rhinoplastie à Paris et peut répondre aux besoins de chirurgie des […]







Grossesse

L’ adoption de certaines précautions après césarienne est essentielle pour protéger la vie de la mère. Il s’agit d’une intervention sûre, mais les risques sont élevés, ainsi que les complications possibles. Avant de vous parler des astuces nécessaires après la césarienne, voyons en quoi consiste une telle intervention chirurgicale. Qu’ est-ce qu’une césarienne ? césarienne est […]



L’hydratation occupe une place de premier plan dans la routine quotidienne de chacun. Durant la grossesse, ses bienfaits s’intensifient, rendant sa consommation non seulement indispensable, mais vitale. L’eau, ce précieux liquide, joue un rôle crucial dans le développement du fœtus, le bon fonctionnement du corps de la mère et la prévention de multiples complications liées […]


Maladie

Naviguer dans le vaste océan des méthodes de contraception peut s’avérer complexe et écrasant. C’est un univers qui s’est considérablement élargi ces dernières décennies, offrant désormais une palette d’options diversifiées pour répondre à une multitude de besoins individuels. Pourtant, malgré cette diversité, plusieurs personnes se sentent souvent perdues dans ce dédale d’informations. Elles cherchent à […]